
While ISO 13485 certification is currently not a requirement in many countries, it is rapidly becoming a de facto worldwide regulatory requirement.The ISO 13485:2016 standard is a globally recognized quality management system (QMS) designed for medical device manufacturers. It provides a framework for companies to demonstrate their commitment to meeting customer and regulatory requirements and ensuring the safety and efficacy of their products. It is also widely used within the Medical Device supply chain (e.g., component manufacturers, cold chain delivery) as many manufacturers like their suppliers to hold this certification.
And perhaps ISO 13485 training needs to be part of your preparation here.
Medical Device Regulations Around the World
In Europe
Regulatory bodies increasingly recognize ISO 13485 certification to demonstrate compliance with international quality and patient/user safety standards.
For example, in the European Union (EU), the Medical Device Regulation (MDR) requires all medical device manufacturers to comply with ISO 13485 certification to demonstrate their compliance with essential safety and performance requirements. The MDR came fully into effect on May 26, 2021, at the end of the transition period, and has significantly increased the scrutiny and requirements for medical device manufacturers. As a result, companies that comply with the MDR and associated standards and associated guidelines maintain access to the EU market, the world's second-largest market for medical devices.
In the USA
Similarly, the United States Food and Drug Administration (FDA) has recognised the ISO 13485 standard to demonstrate compliance with its Quality System Regulation (QSR) for medical devices. The FDA has stated that it expects medical device manufacturers to be certified to ISO 13485 by an accredited third-party certification body. Failure to comply with the QSR can result in fines, product recalls, and loss of market access.
Other Countries
Countries such as Australia, Canada, and Japan have also recognised ISO 13485 certification to demonstrate compliance with their respective regulatory requirements. Indeed, compliance with the standard is mandatory in some countries, such as Canada and Australia, and is often a requirement for companies seeking to market their products in other countries. Furthermore, as medical device markets expand globally, more countries will likely adopt ISO 13485 certification requirements.
Continued alignment of UK Regulations with those of the EU is expected to continue (as anything else would be economic suicide). As with the EU, ISO 13485 will remain the relevant QMS standard for medical device manufacture.

MDSAP incorporates ISO 13485 Requirements
And then there is the MDSAP Scheme. The Medical Device Single Audit Program (MDSAP) allows medical device manufacturers to undergo a single audit that satisfies the regulatory requirements of multiple countries. The program was developed by the International Medical Device Regulators Forum (IMDRF), a group of regulatory bodies worldwide.
Under the MDSAP, manufacturers can undergo an audit by an authorized auditing organization that assesses their compliance with the regulatory requirements of participating countries, including Canada, Australia, Japan, Brazil, and the United States. The audit covers aspects such as QMS, regulatory, and product-specific requirements.
The MDSAP aims to streamline the regulatory process for medical device manufacturers, reduce the regulatory burden on manufacturers, and improve the consistency of regulatory requirements across participating countries. Compliance with the program is voluntary but is becoming increasingly popular as more countries participate. And certification to ISO 13485 is a prerequisite of MDSAP.
Not just a Regulatory Requirement
ISO 13485 certification is not only becoming a regulatory requirement, but it also offers significant benefits for medical device manufacturers. Certification can help companies improve their QMS, reduce costs, and enhance customer satisfaction. The standard provides a framework for companies to establish and maintain an effective QMS, including processes for design and development, risk management, and quality control. In addition, certification demonstrates to customers and stakeholders that a company is committed to meeting international quality and safety standards.

ISO 13485 Internal Auditor Course image map. Click on the image thumbnail to see the table in full size or the button on the other side to see the full overview of the course.
Is ISO 13485:2016 not due for a Revision?
Under the usual ISO Rules for ISO management system standards, this 2016 standard should be reviewed and revised within ten years. A revision was initiated back in 2019 but later quietly dropped. The problem is that over the past seven years or so, there have been many significant changes and proposed changes, to medical device regulations, especially in Europe and the USA (not to mention Brexit). A new revision only makes sense after major regulatory changes have been completed and ‘bedded in’. So a Revision before 2030 seems most unlikely.
The Road Ahead for ISO 13485:2016
So, ISO 13485:2016 certification is rapidly becoming a worldwide regulatory requirement for medical device manufacturers. Certification helps companies comply with regulatory requirements and provides significant benefits, including improved QMS, reduced costs, and enhanced customer satisfaction. In addition, as the global medical device market expands, more countries will likely adopt ISO 13485 certification requirements. Therefore, medical device manufacturers should consider obtaining ISO 13485 certification to remain competitive in the global market and demonstrate their commitment to quality and safety.
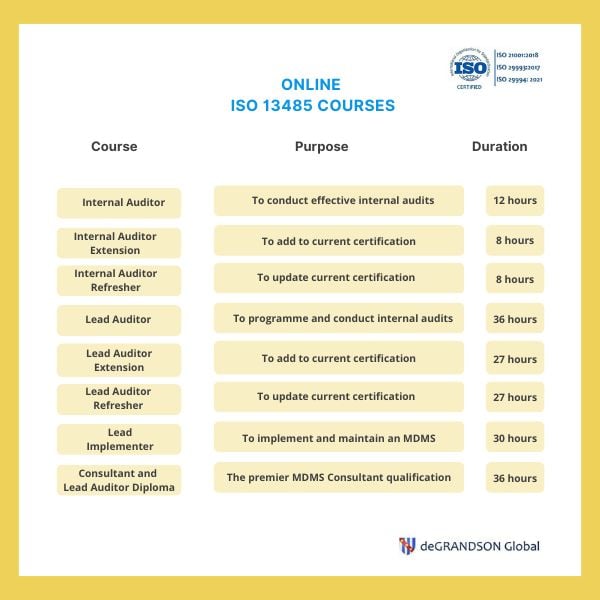
ISO 13485 image map. Just click on any course you are interested in to see their individual overviews.
Related Articles
- EU MDR and IVDR Guidance on PRRC Requirements
- ISO 13485: Critical Subcontractors and Crucial Suppliers
- IVDR Transition Deadline extended up to 2027 – reality bites
deGRANDSON Global is an ISO Certified Educational Organization
In October 2021, we secured certification to three education-related ISO Standards. As a result, we now have a university-grade management system conforming to the requirements of …
October 2021, we secured certification to three education-related ISO Standards. As a result, we now have a university-grade management system conforming to the requirements of …
We have chosen ISO 21001 certification because, unlike IRCA and Exemplar badges (which we believe are commercially compromised), it is based on independent third-party assessment. In addition, it is a ‘university grade’ standard in use globally by schools, colleges, and universities to demonstrate their competence.